Bird or avian malaria is caused by Plasmodium and Haemoproteus parasites – different species than those affecting humans. However, strictly speaking, only disease caused by Plasmodium infection is referred to as avian malaria. Plasmodium and Haemoproteus belong to the haemosporidian (Phylum: Apicomplexa) group of parasites. These blood-borne parasites are transmitted from one bird to another via vectors— mosquitoes transmit Plasmodium whereas biting midges and louse flies transmit Haemoproteus. For my PhD research, I studied avian malaria parasites from songbird communities in a global biodiversity hotspot, the Western Ghats mountains in southern India.
1) Why some parasites emerge in novel communities while others do not?
Avian haemosporidian parasites are known to be generally less harmful to their hosts they do have negative effects on birds’ survival/reproduction and fitness. Large scale mortalities have occurred in Hawaiian and New Zealand island bird populations due to the introduction of Plasmodium parasites but no such mortalities have been reported for Haemoproteus.
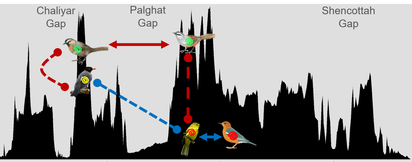
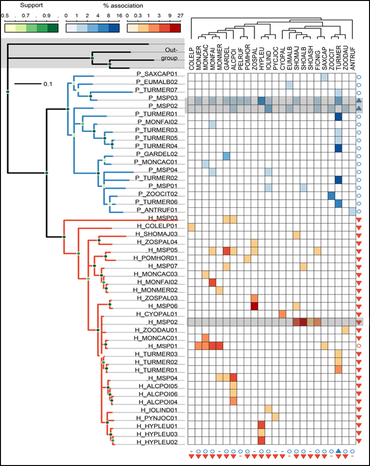
Could there be some ecological and evolutionary differences between Plasmodium and Haemoproteus that influences the increased likelihood of emergence of Plasmodium but not Haemoproteus? Could this be related to the ability of some parasites to rapidly switch to novel hosts and emerge to spread to new locations while others are geographically restricted? This was one of the questions that we tackled in this project. Our research showed that Plasmodium and Haemoproteus differed in their infection dynamics. Haemoproteus were more common than Plasmodium in the Western Ghats Sky Island bird community. |
Gupta et al. 2019, Proc. R. Soc. B
Plasmodium parasites were host-generalists—capable of rapidly jumping across multiple bird species whereas Haemoproteus were host-specialists and restricted to a few bird species. Community structure of Haemoproteus was more affected by host phylogeny/ecology whereas Plasmodium were affected by geographic distance, potentially leading to greater likelihood of emergence by Plasmodium in novel host communities.
|
2) Why are some bird species more infected by blood parasites than others?
Understanding why some host species are more infected than others is critical from wildlife health and conservation perspective as the number and severity of emerging infections are increasing globally. Identifying the ecological and evolutionary factors that shape variation in infection risk among host species and influence potential spillover risk to naïve hosts is needed to understand infectious disease dynamics in wildlife populations.
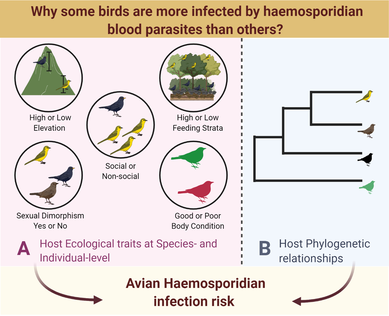
Many factors operating at the host species/individual level can affect avian haemosporidian infection risk in bird communities via increased exposure to parasites. For example, host ecology, host life history traits, behavior can all affect exposure of hosts to parasites/more vectors and increase their chances of getting infected. These traits can include species elevation, foraging strata, sociality, sexual dimorphism, or individual body condition. |
Gupta et al. 2020, Parasites & Vectors
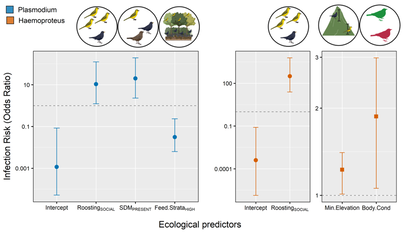
Additionally, phylogenetic relatedness of the hosts can also affect hosts susceptibility to infection. In this project, we tested whether host ecological factors and evolutionary history affect variation in avian haemosporidian infection risk in the Western Ghats. Our study showed that host ecological traits and host phylogeny differentially influence infection risk by Plasmodium and Haemoproteus. We found that infection risk among bird communities was affected by host ecological traits promoting parasite exposure (e.g., sociality, foraging strata) and traits associated with host susceptibility (e.g., sexual dimorphism, body condition). Additionally, host phylogeny contributed substantially in predicting Haemoproteus infection risk compared to Plasmodium, reiterating that host ecology and host phylogeny together influence infection dynamics. |
Does anthropogenic fragmentation impact genetic connectivity?
|
This project is one of the first projects that kickstarted my research career and is still close to my heart. Here, we examined the population genetic effects of natural and anthropogenic fragmentation on two sky-island-endemic birds, belonging to genus Sholicola, in the Western Ghats. The deep valleys and the mosaic of forests and grasslands in the Western Ghats sky-islands create natural patchiness while the loss of approximately 85% of the habitat due to anthropogenic modifications has caused additional patchiness. To assess genetic connectivity, we generated population level genetic data from 218 individuals using microsatellite markers. We showed that fragmented habitats had reduced population genetic connectivity with the timing roughly corresponding with when deforestation occurred.
|
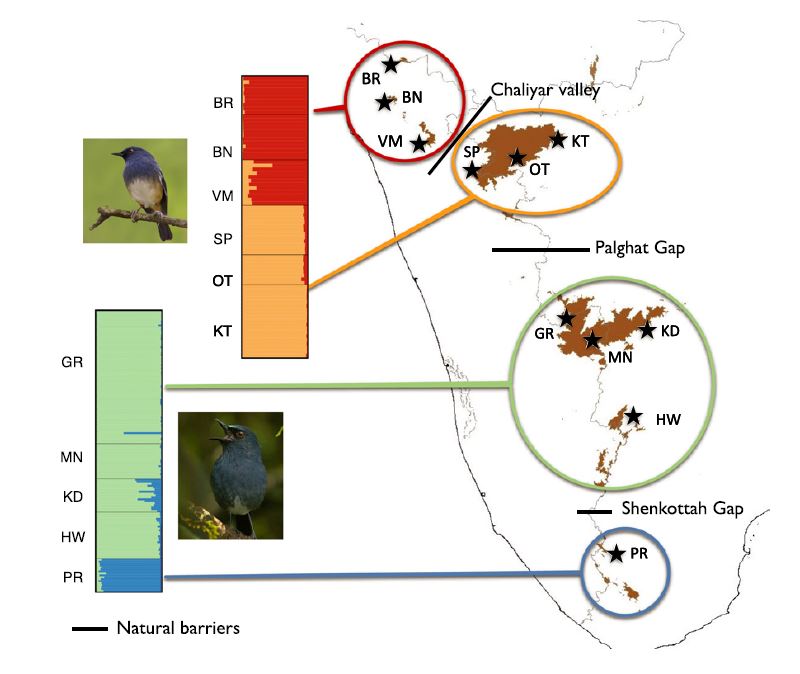
|
Do topographic valleys impact genetic differentiation across the montane bird community in India?
|
In continuation with the project on Sholicola spp. genetic connectivity, this project aimed at understanding comparative phylogeography patterns across the entire tropical sky-island bird community (comprising montane specialists and widespread generalists) in the Western Ghats, India. I was fascinated by this system as these sky-islands harbour exceptionally high levels of endemism within the Western Ghats biodiversity hotspot and are geographically isolated by topographic valleys. Such valleys can act as bridges or barriers for gene flow, depending on the degree of specialization of species.
To examine genetic divergence in different species, we analyzed a genetic dataset from 23 species using a comparative phylogenetic approach. Our results revealed that ten out of 23 species were genetically differentiated due to topographic valleys. The lack of genetic differentiation for montane specialists was quite surprising and on-going gene flow, or extinction-recolonization dynamics may potentially explain this pattern. |
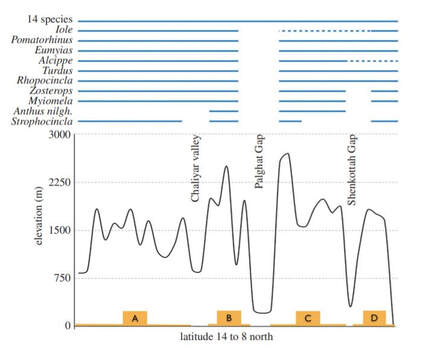
|
A novel target enrichment bait set to illuminate genome-scale phylogeny of Plasmodium and related haemosporidian blood parasites
|
Haemosporidians (Phylum: Apicomplexa, Order: Haemosporida) are a diverse group of vector-borne protozoan parasites that infect numerous vertebrate hosts, including mammals, birds and reptiles. Due to its diversity and role as a causative agent for human malaria, many studies have primarily focused on Plasmodium spp. infecting primates and rodents, and consequently, large-scale genomic data for other haemosporidian parasites such as those infecting birds is lacking. Subsequently, evolutionary origins of avian haemosporidians, mechanisms of co-evolution and speciation leading to its exceptional global diversity and biogeography of these parasites is less well understood.
Recent advancements in NGS technologies and parallel development in bioinformatics tools have now made it easier to generate data rapidly and reliably from a reproducible subset of the genome, scalable to a large number of individuals using reduced representation technologies. One class of techniques that have received huge attention are DNA sequence capture or target enrichment, which allows one to generate data from thousands of targeted markers from hundreds of individuals. These are especially useful for assessing evolutionary relationships at both deep and shallow scales, thus allowing us to answer phylogenetic and population genetic questions. Sequence capture approach based on highly conserved genomic elements [ultraconserved elements (UCEs)] have become quite popular in the last decade due to their wider taxonomic phylogenetic applicability. UCE-based sequence capture approach is especially advantageous as it can be used to obtain genomic data from degraded DNA samples is thus useful for obtaining parasite genomic data in cases where host contamination has posed challenges for obtaining genomic data and fewer genomic resources are available. |
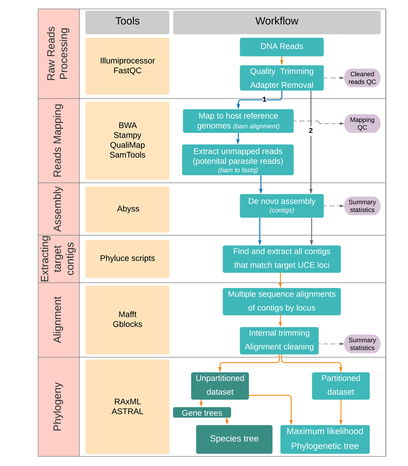
Bioinformatics workflow for Phylogenomic analysis
With this background in mind, we developed a novel UCE probe set targeting conserved loci across representative Plasmodium and Haemoproteus genomes. We tested the utility of DNA sequence capture approach to selectively enrich parasite DNA in eight clinical samples and 32 wild bird samples infected with haemosporidian parasites. We then used this parasite phylogenomic data from ~300 genome-wide UCE loci to establish a robust phylogeny for avian haemosporidians and related malaria parasites. Our results provide the most comprehensive phylogenomic dataset to date for avian and other related haemosporidian parasites and likely challenges the current taxonomic relationships of avian haemosporidians.
Phylogenomic analysis for this project is in progress. Please check back later for updated results. |
I am looking to expand this project and estimate a more robust haemosporidian phylogeny with a broader set of infected bird/bat/ungulate samples. Please reach out to me if you would like to collaborate on this work.
